

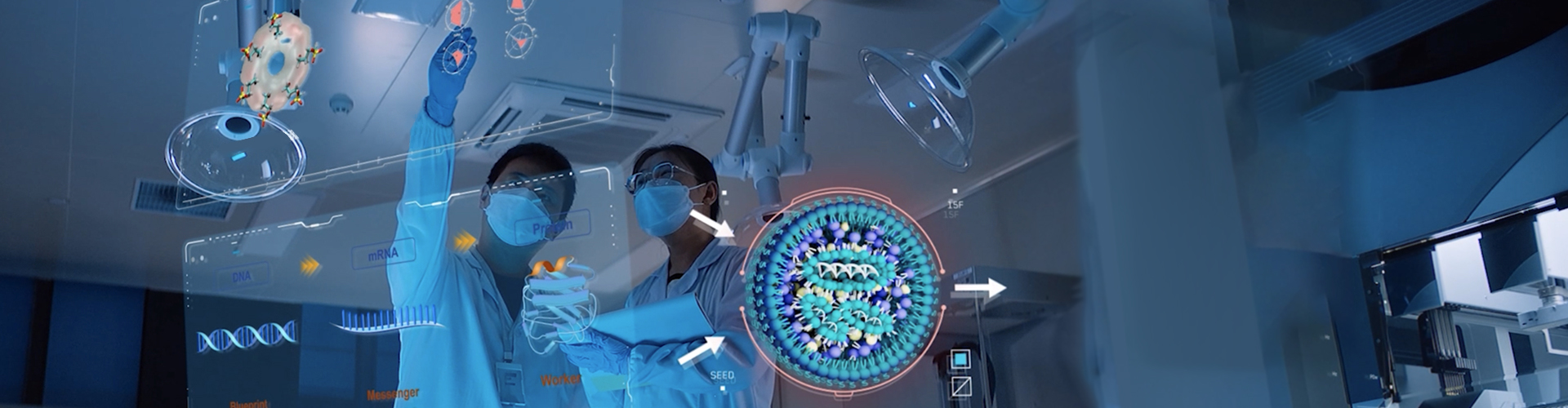

Understanding interactions between nanomaterials and biological molecules — such as proteins and nucleic acids — is critical to ensuring the efficiency, precision, and safety of active agent delivery. Our AiLNP platform applies advanced computational modeling to simulate these interactions and predict the behavior of supramolecular assemblies. At its core, AiLNP leverages our proprietary lipid library, lipid language models, and generative algorithms to identify and design optimal lipid candidates. Predictive AI and molecular dynamics models provide mechanistic insights and predict over 20 lipid properties. The platform enables systematic design and optimization of LNP components — including ionizable lipids, helper lipids, cholesterol, and PEG-lipids — and their ratios to create tailored, high-performance formulations.
Through our DATALOTS high-throughput platform, in-house animal models, and AI-guided screening workflows, AiLNP accelerates in vivo data collection and optimization. It also supports rapid, scalable, and GMP-compliant LNP synthesis, streamlining the path from discovery to development.
The complexity of drug delivery increases significantly when supramolecular structures like LNPs interact with living systems, and when biomolecules such as mRNA engage with the cellular machinery. A deep understanding of these dynamic, multiscale interactions is critical for precise biological modulation. Our integrated AiLNP and AiRNA platforms are built to meet this challenge.
AiLNP and AiRNA platforms enable AI-driven, full-length mRNA sequence design optimized for expression, protein folding, stability, and manufacturability. Using generative AI, the platforms also design organ-targeting untranslated regions (UTRs), validated through highthroughput cell-based UTR screening datasets. Additionally, they simulate and predict key in vivo behaviors — including biodistribution, cellular uptake, endosomal escape, and downstream biological responses — of both LNPs and mRNA. These predictive models are continuously refined with experimental data, enabling the co-optimization of lipid components, formulations, and mRNA sequences to function synergistically and maximize therapeutic efficacy.

AiProtein adopts METiS's proprietary "dry lab + wet lab + agent" innovation paradigm, serving as a design platform focused on proteins, antibodies, and other macromolecules.
The AiProtein platform encompasses self-developed models and algorithms including de novo generative models, language/prediction models, affinity prediction models, druggability prediction models, and active learning-driven dry-wet iterative algorithms, with technical integration in nanobody engineering. Additionally, METiS has developed AARON, a next-generation antibody design agent capable of orchestrating various AI models developed by METiS. AARON provides scientists with a visual interactive interface, enabling integrated operations for antibody generation, property prediction, affinity optimization, and dry-wet closed-loop workflows.

In small molecule drug discovery and development, our AiTEM platform is at the forefront of understanding nanoscale interactions between chemical molecules and biological systems — interactions that are critical to formulation success. AiTEM leverages advanced AI, high-throughput iteration, molecular dynamics, and quantum chemistry to power proprietary algorithms that design and optimize drug formulations, including excipient types and ratios. It uses quantum chemistry and molecular dynamics simulations to predict drug — excipient interactions and identify optimal excipient candidates.
The platform also enables optimization of nanoscale formulations such as cosolvent solvation, micelles, solid dispersions, cyclodextrin encapsulations, and microspheres to improve solubility and bioavailability. Formulation samples can be rapidly tested using automated high throughput platforms. AI-guided iteration further analyzes experimental outputs — including solubility, stability, and permeability — to recommend top-performing formulations for further development.
